Die Oxidschicht hemmt damit sowohl die anodischen Reaktionen (Metallauflösung), als auch die kathodische Reaktion (in der Regel ist das die Reduktion von im Elektrolyten gelösten Sauerstoff). Die Veränderung des Elek- trodenpotentials bewirkt lediglich eine Änderung der Schichtdicke der Sperrschicht.
Fließt ein noch messbarer Reststrom, so handelt es sich um Reaktionen an Fehlstellen der Schicht. Ihr Flächenanteil an der Elektrodenfläche beträgt θ = 10-3–10-5 [1]. Das Zeichen θ (Theta) wurde in Anlehnung an den Bedeckungsgrad, z.B. mit Inhibitoren, gewählt.
Solche Fehler, Poren im Oxidfilm, heilen in der Regel schnell selbst wieder aus. Bei einem Elektrodenpotential unterhalb von etwa -1000 mVH tritt kathodische Korrosion infolge Alkalisierung der Oberfläche durch Wasserzersetzung auf. Oberhalb dieses Potentials hängt die Summen-Stromdichte vom 02-Gehalt und vom pH-Wert der Elektrolytlösung ab.
Reinaluminium ist z. B. in Sulfatlösung ungefährdet (der Widerstand der Passivschicht ist sehr hoch). Enthält die Lösung aber Chlorid, so tritt Lochfraß ein. Das Chlorid führt zu Fehlstellen in der Passivschicht.
Nach
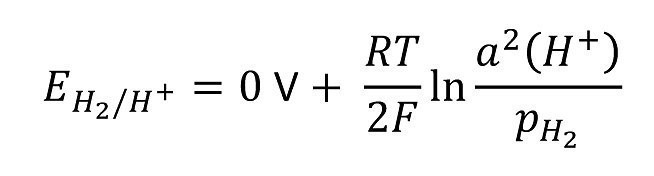
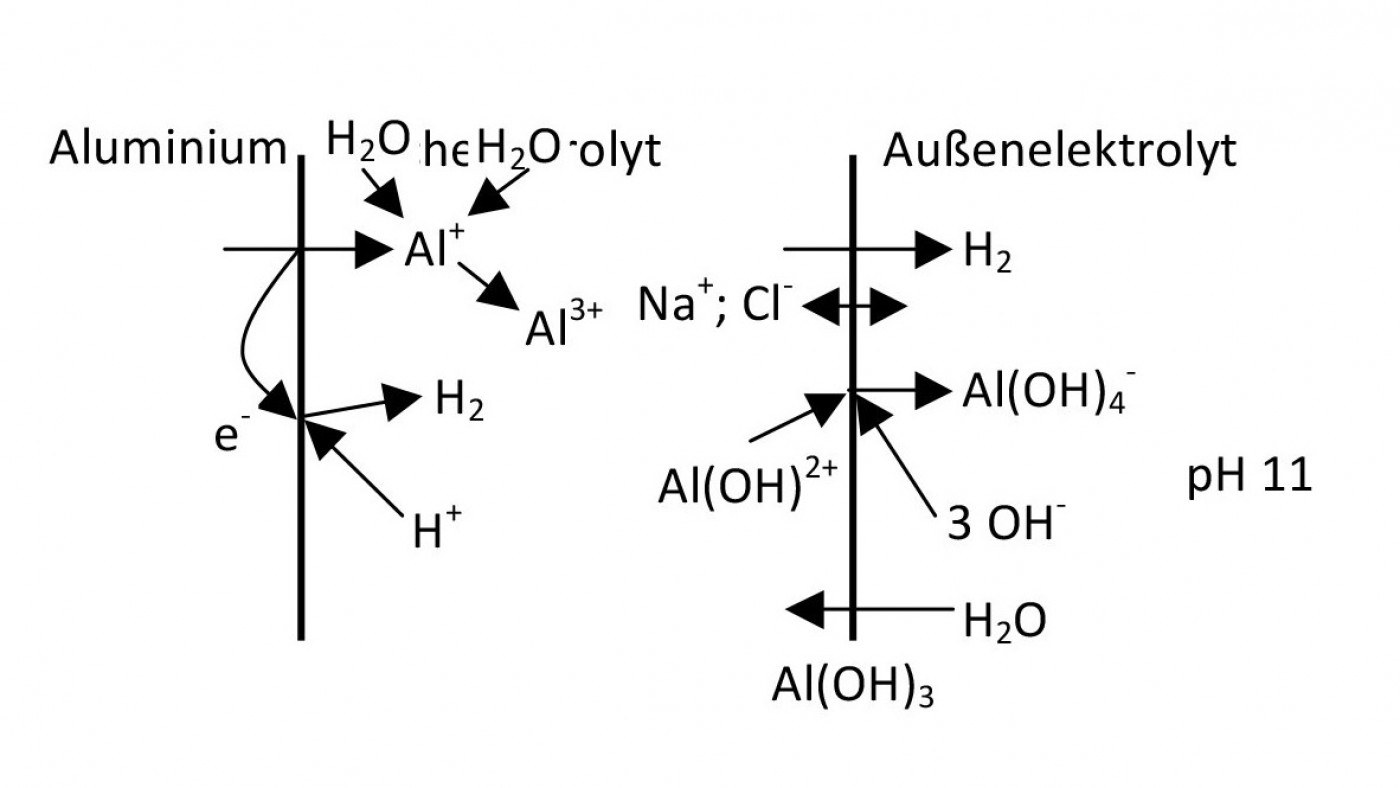
ist das Potential der Wasserstoffabscheidung aus dem angesäuerten Lochelektrolyten positiver als das der Abscheidung aus dem äußeren Elektrolyten.
Die Aluminiumionen [Al(H2O)6]3+ wirken bereits als schwache Säure (pKs = 4,97, [2]):
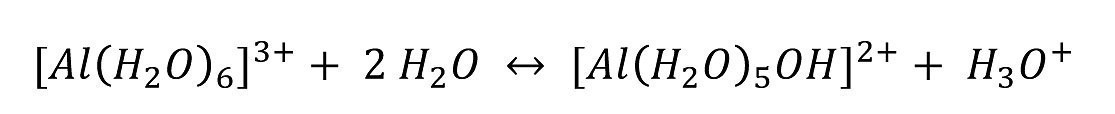
Aluminium kann in drei Stufen hydrolysieren. Die Stufen werden natürlich nacheinander durchlaufen.
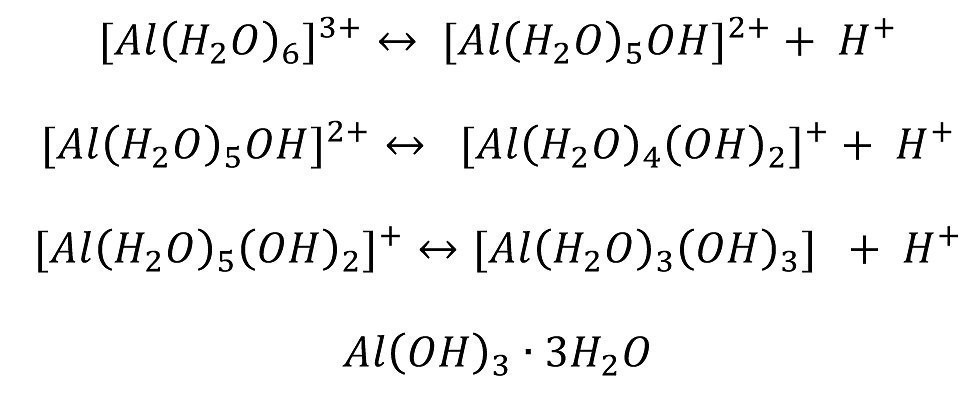
In alkalischer Lösung
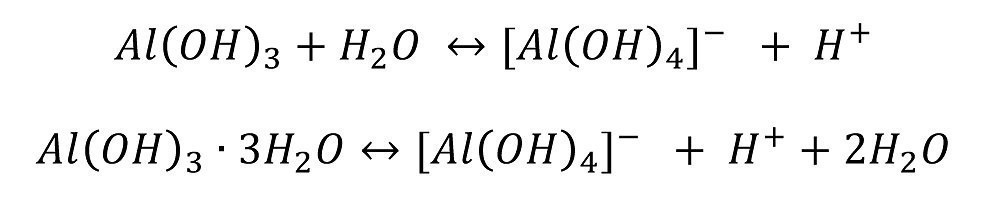
Nach Kaesche ist der pH-Wert nach der ersten Hydrolysestufe schon so hoch (bzw. niedrig), dass aus einer gesättigten AlCl3-Lösung kein Al(OH)3 mehr ausfallen kann (Abb. 1).
Die Lochfraßstellen wachsen etwa halbkugelförmig. Allerdings nicht glatt. Da sich auch im Loch auf der Oberfläche immer wieder eine Oxidhaut bildet, muss diese immer wieder durchbrochen werden. Das ist bei einer genügenden Chloridkonzentration auch der Fall. Anderenfalls kann das Loch durch diese Oxidschicht selbst heilen.
Der Chloridgehalt wirkt auf die Austauschstromdichte der Aluminiumauflösung. Wieso steigt die Austauschstromdichte mit dem Chloridgehalt an?
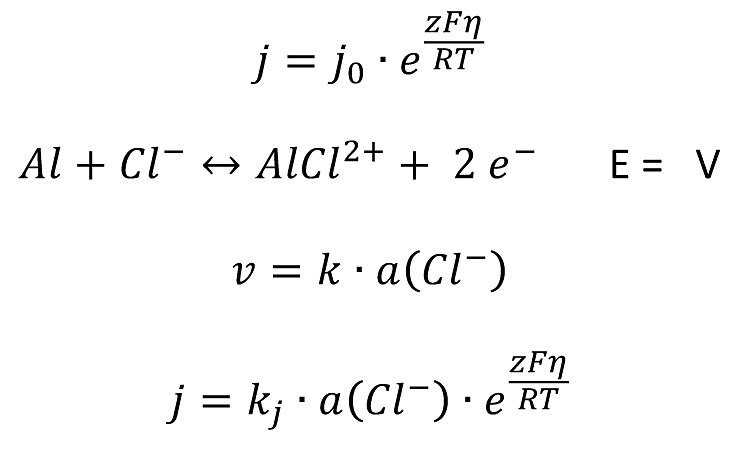
Lochfraßstromdichte-Potential-Kurve
Ausgehend von der Polarisationskurve
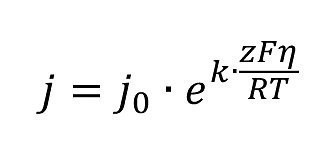
erhält man mit dem hohen Anteil Widerstandspolarisa- tion ηR im Loch.
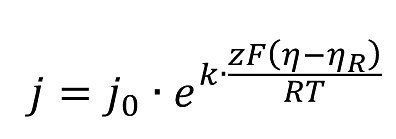
Kaesche: S. 320: Es fällt auf, dass beim Lochfraßpotential selbst die aus dem Wachstum der Lochfraßstellen selbst ermittelte Stromdichte IL der Aluminiumauflösung für alle Chloridkonzentrationen gleich wird, nämlich, auf die Lochmündung bezogen 0,6 A/cm2 (62 A/dm2).
Das Lochfraßpotential selbst variiert mit der Chloridkonzentration. Das heißt aber andererseits, die Chloridkonzentration geht mit in die Nernstsche Gleichung ein und das Chlorid ist direkt an der Durchtrittsreaktion beteiligt.
Anodische Geschwindigkeitskonstante
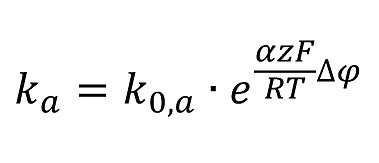
Stromdichte
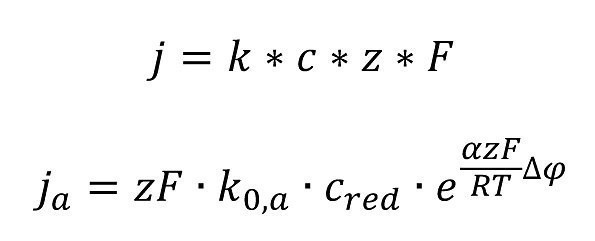
Gleiche Austauschstromdichten funktionieren nur, wenn die Aluminiumchloridlösung im Loch immer gesättigt ist. Das ist bei der hohen Stromdichte aber leicht (ca. 3,375 mol/L Wasser) möglich.
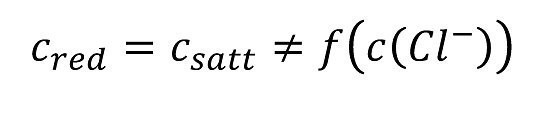
Mit Widerstandspolarisation (= Messfehler)
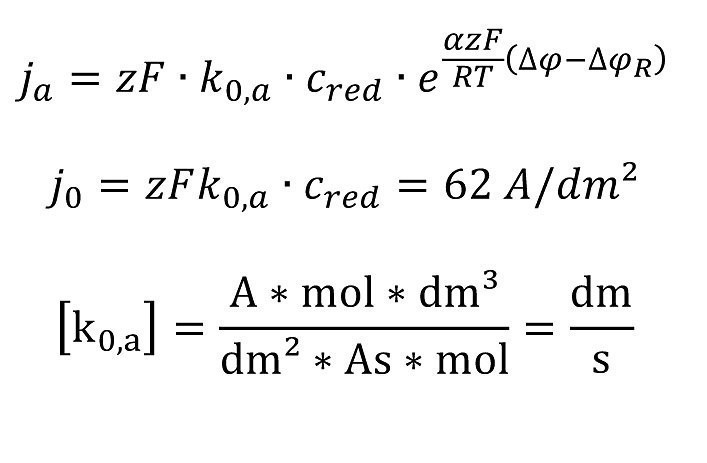
Mit xred: und x= 0,1579
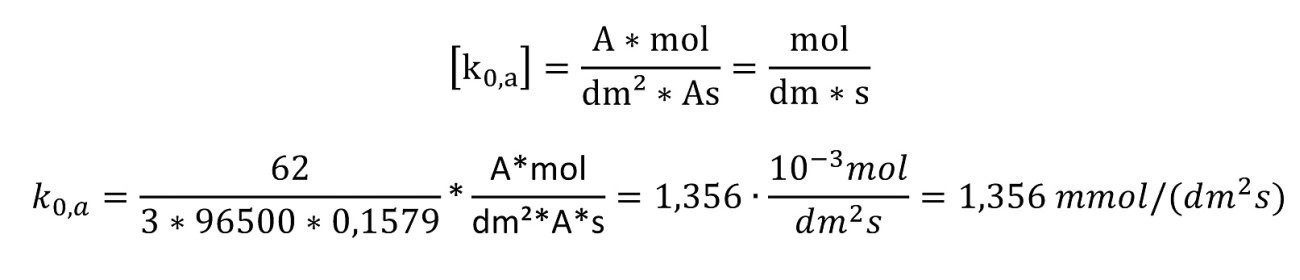
D = 0,0613*10-9 m2/s für Al3+ in Wasser
Diffusionsstromdichte

Mit zunehmender Zeit muss demnach die Auflösungsstromdichte geringer werden, weil sich die Länge des Widerstandswegs vergrößert. Die Rauheit im Loch ist nicht berücksichtigt.
Wachstum: t3-Gesetz?
vL = 0 bei E = EL und vL > 0 bei E > EL und vL↑,
wenn c(Cl-)↑
halbkugeliges Wachstum – Diffusionsform?
So, wie eine Aluminiumelektrode fluoridsensitiv ist,
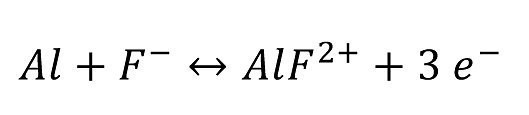
ist sie offensichtlich auch Chlorid-sensitiv.

Das heißt aber, mit zunehmendem Chloridgehalt wird die Elektrode negativer:
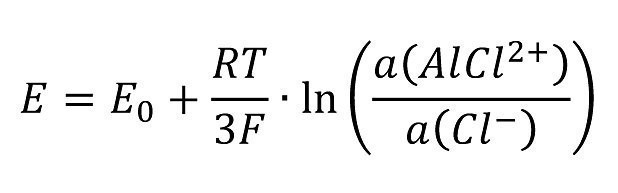
Und zwar um knapp 20 mV je Größeneinheit der Chloridkonzentration.
Al+ wird in manchen elektrochemischen Reaktionen postuliert. Das ist leicht nachvollziehbar, wenn man davon ausgeht, dass immer nur zwei Teilchen zusammenstoßen. Es muss also Zwischenschritte der Summenreaktion geben. Die Eigenschaften der niederwertigen Ionen sind aber nicht messbar.

Aus ΔG = -878 kJ/mol ergibt sich für AlCl3 mit

z =3 und F = 96486 As/mol
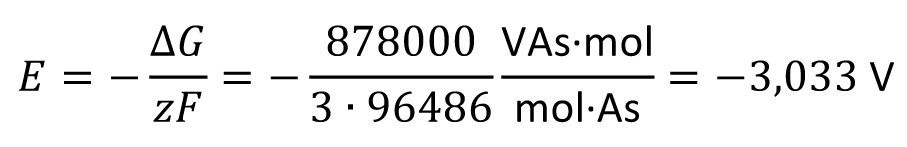
Aus ΔG = -1322 kJ/mol ergibt sich für AlF3 mit
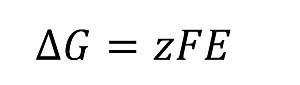
z =3 und F = 96486 As/mol
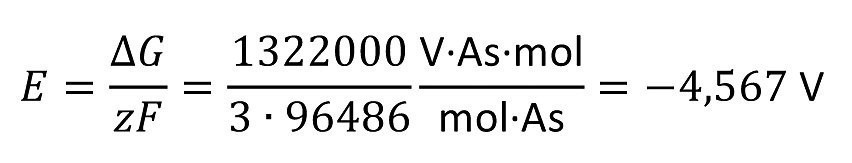
Für die Zelle der Lochfraßkorrosion mit Chloridlösung ergibt sich
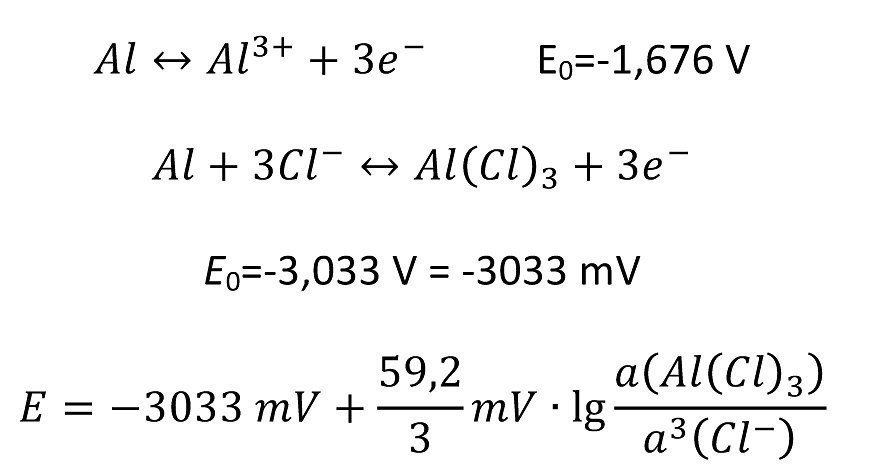
Teilt man die Gleichung auf

und geht davon aus, dass die Lösung am Boden des Loches ständig an Aluminiumchlorid gesättigt ist [3], so ergeben sich für den Term des Aluminiumchlorids etwa 10 mV (wenn man den sehr niedrigen Aktivitätskoeffizienten nicht berücksichtigt), d.h.
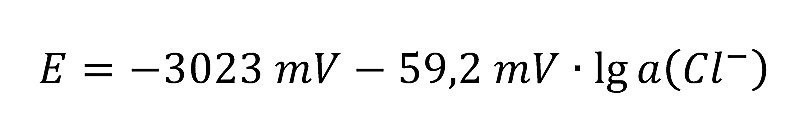
Die Aussage, dass sich die Abhängigkeit des Lochfraß- potentials vom Chloridgehalt erklärt, ist falsch! Richtig ist: So erklärt sich, dass das Lochfraßpotential unabhängig vom Chloridgehalt der Lösung ist.
Für die Stromdichte-Potential-Kurve ist sie aber doch wichtig. Je höher die Chloridkonzentration, umso steiler ihr Anstieg. Die Überspannung im Exponenten verändert sich.
| 4 | mol/L | -35,642 | mV |
| 1 | mol/L |
±00,000 |
mV |
| 0,25 | mol/L | +35,642 | mV |
Ohmsches Gesetz im Loch
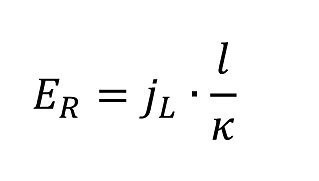
Vertiefung
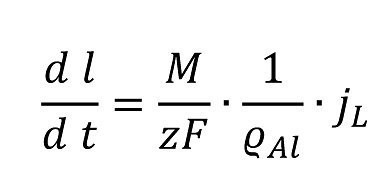
So erhält man ein scheinbar lineares Zeitgesetz

Ersetzt man die Stromdichte
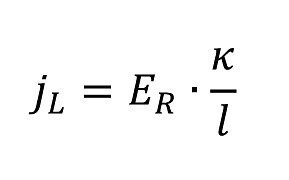
so ergibt sich (bei konstantem ohmschen Spannungs- abfall)
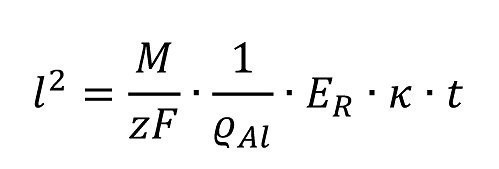
das heißt, ein Wurzelgesetz.
Diffusion

Geometrische Verhältnisse
Bei einer runden Lochfläche (AL = πr2), die halbkugelig hinterfressen ist, ist die Oberfläche der Halbkugel AK = 2πr2. Folglich verhält sich die Auflösungsstromdichte an der Kugelwand zur Lochstromdichte wie 1:2. Das gilt unabhängig von der Rauheit der Halbkugeloberfläche. Die wahre Stromdichte ist wesentlich geringer, weil der Lochboden sehr ungleichmäßig korrodiert. Häufig besitzen diese Vertiefungen die Form von Ätzgrübchen, d.h. einzelne Kristalle werden herausgelöst.
Eine bevorzugte Ätzrichtung ist die <100>-Richtung. Übergang vom Ätzen zum Elektropolieren beseitigt diese Auflösungsform.
Manche Ätztunnel sehen nach der Präparation im Elektronenmikroskop aus wie Kristalle im feldorientierten Texturtyp nach Fischer, nur dass es keine Kristalle sind, sondern die Spuren von Kristallen. Man hat die Lochfraßtunnel präpariert, indem man sie nachträglich anodisiert und das Aluminium weggelöst hat.
Praktische Anwendung findet der Fehler des Lochfraßes bei der gleichmäßigen Aufrauhung des Aluminiums mit Salzsäure.
Die Untersuchungen der Auflösegeschwindigkeit des Aluminiums führen immer wieder auf Stromdichten zwischen 10 A/cm2 und 100 A/cm2. Und das bei einer auf jeden Fall vorhandenen Deckschicht. Abgesehen von der natürlichen Oxidschicht bilden sich bei so hohen Stromdichten auf jeden Fall Salzschichten. Diese Erfahrung hat jeder gemacht, der versucht hat, Stromdichten von 20 A/dm2 und mehr mit löslichen Anoden zu realisieren. Diese sich ausbildende anodische Schicht ist wieder doppellagig, wobei die äußere Schicht einen ohmschen Widerstand darstellt, d.h. es handelt sich um hydratisierte Ionen. Die porenfreie innere Lage leitet den Strom durch Ionentransport mit exponentieller „Hochfeld“-Spannungsabhängigkeit.
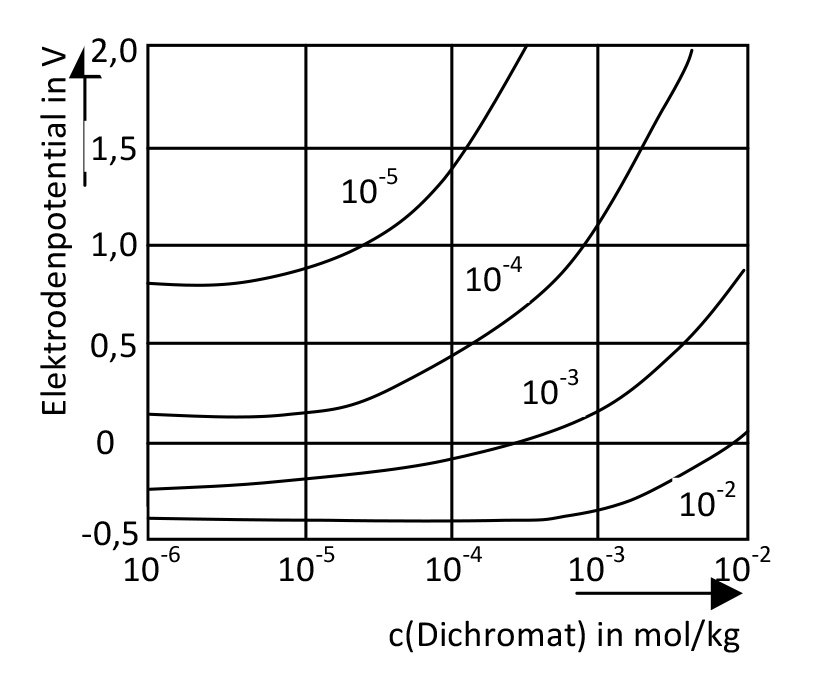
 in mol/kg. Die potentiodynamisch mit 1 V/h aufgenommenen Kurven charakterisieren das Potential des Einsetzens des schnellen Lochfraßes. Die Inhibitormoleküle werden stärker an der Aluminiumoberfläche adsorbiert als die Halogenidmoleküle") Abb. 2: Wirkung von Dichromationen in chloridhaltigen Lochfraß-Korrosionselektrolyten; Parameter: c(Cl-) in mol/kg. Die potentiodynamisch mit 1 V/h aufgenommenen Kurven charakterisieren das Potential des Einsetzens des schnellen Lochfraßes. Die Inhibitormoleküle werden stärker an der Aluminiumoberfläche adsorbiert als die Halogenidmoleküle
Abb. 2: Wirkung von Dichromationen in chloridhaltigen Lochfraß-Korrosionselektrolyten; Parameter: c(Cl-) in mol/kg. Die potentiodynamisch mit 1 V/h aufgenommenen Kurven charakterisieren das Potential des Einsetzens des schnellen Lochfraßes. Die Inhibitormoleküle werden stärker an der Aluminiumoberfläche adsorbiert als die Halogenidmoleküle
Bei der Auflösung in alkalischen Elektrolyten läuft die Reaktion auf jeden Fall über das feste Oxid bzw. Hydroxid. In stärker alkalischen Elektrolyten wird angenommen, dass es sich um Hydrargillit handelt und in schwächer alkalischen Elektrolyten um Bayerit [4]. Dieser Reaktionsteilschritt soll der Geschwindigkeits-bestimmende sein [3, 4]. Dabei wird die Auflösegeschwindigkeit durch die Diffusion von OH- und [Al(OH)4]- gesteuert [4]. Das Lochfraßpotential wird in der Reihe Cl-, Br-, I- positiver. Es wird als Schwelle der Aktivierung der Keimbildung der Ätzgrübchen verstanden.
Inhibition des Lochfraßes
Als Inhibitoren wirken u.a. Chromationen. Anderson und Hocking [5] zeigten, dass es dabei nicht auf die Konzentration der Inhibitormoleküle im Korrosionselektrolyten ankommt, sondern auf ihr Verhältnis zu den Passivitäts-aufhebenden Ionen, z. B. Chlorid. Der Beginn des schnellen Lochfraßes ist in diesem Zusammenhang zwar deutlich von der Chromatkonzentration abhängig (Abb. 2), das stationäre Lochfraßpotential jedoch kaum.
Literatur
[1] Ergang, R.; Masing, G., Z. f. Metallkunde, 41(1950), 272
[2] https://hps.hs-regensburg.de/~heh39273/aufsaetze/lfr1.pdf
[3] Kaesche, H.Z.: Physikalische Chemie NF 34(1962), 87 und Kaesche, H.Z.: Werkstoffe und Korrosion 14(1963), 557
[4] Heusler, K.E.; Allgeier, W.: Werkstoffe und Korrosion 22(1971), 297
[5] Anderson, P.J.; Hocking, M.E.: J. Appl. Chem. 8(1964), 352




