
... as an explanation for inhomogeneous galvanic metal deposition and metal dissolution
In 1991, the journal Galvanotechnik [1] discussed the problem of the formation of distinct "bright spots" during pure gold deposition, which not only differed visually from the surrounding areas, but also the layer thickness in the spots and the respective surroundings was different.
In his reply, the expert questioned pointed out that these structural differences in terms of area could be related to locally different inhibition by brighteners and correspondingly different local current densities, and that similar findings have also been observed with tin-lead layers on printed circuit boards and copper and nickel deposition. As the most probable cause for the varying degrees of inhibition and the associated differences in layer thickness, the expert cited the flow plastics already described by Fischer [2] and Matschke [3], whereby, interestingly, the expert assigned an S-shaped current density-voltage characteristic to this phenomenon. However, the spot-like structure discussed did not necessarily speak for a phenomenon caused by hydrodynamics, since, as the expert showed in his contribution using a flow plastic itself as an example, elongated, groove-like metal deposits tend to occur.
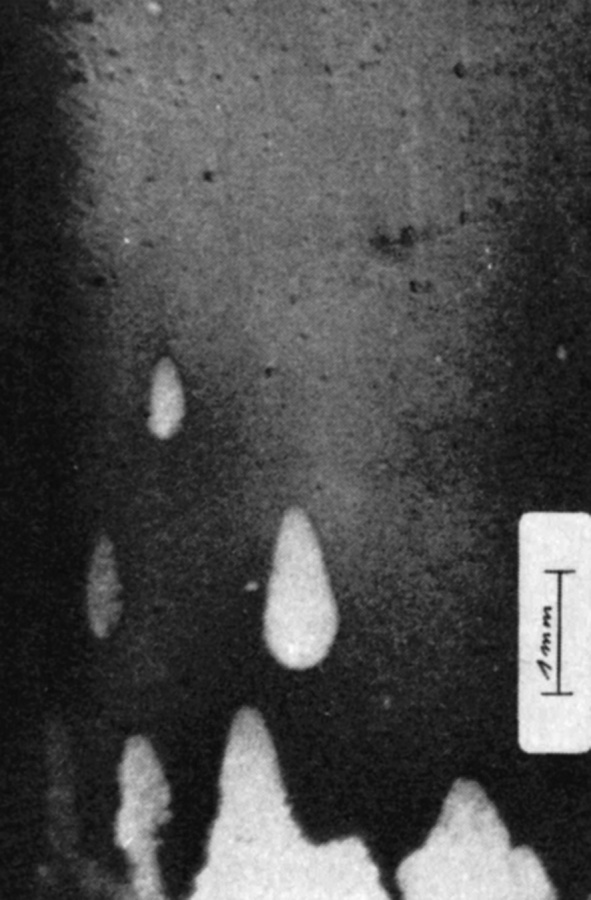
![Abb. 1 links: Kathodenoberfläche mit koexistierenden Zuständen bei Unterdosierung von Chlorid [4]](/images/stories/Abo-2022-11/gt-2022-11-0043.jpg "Abb. 1 links: Kathodenoberfläche mit koexistierenden Zuständen bei Unterdosierung von Chlorid [4]") Fig. 1 left: Cathode surface with coexisting states with underdosing of chloride [4]
Fig. 1 left: Cathode surface with coexisting states with underdosing of chloride [4]At that time, however, the expert would have had the opportunity to include in his expertise the theory of "Equally conditioned coexisting stationary states" published by Osterwald and his colleagues 1 ½ decades ago, which could have better explained the different metal precipitations that occurred side by side during gold deposition. As early as 1976, Osterwald and Schulte [4] published images of electrodeposited copper surfaces in the journal Galvanotechnik that were very similar to the "spots" discussed above (Fig. 1) and whose occurrence was accompanied by a strikingly steep drop in the potentiodynamic current density-voltage characteristic curve during copper deposition with the organic additive polyethylene glycol and chloride additive (Fig. 2).
These observations by Osterwald and Schulte were made at a time when the Berlin Institute of Metallurgy was interested in the reasons for the inhomogeneous metal deposition of electrodeposited layers using organic additives and recognized very different causes for the formation of inhomogeneous surface structures, which were explained and experimentally substantiated by different models, such as [I] a new leveling theory based on the surface-controlled mechanism [5, 20], [II] hydrodynamically induced periodic groove structures [6, 19] and [III] the theory of coexisting steady states for the formation of non-uniform surface structures on metal surfaces during metal deposition and dissolution [4, 7, 9, 10, 18].
![Abb. 2 rechts: Potentiodynamische StromdichteÜberspannungskurve für die Kupferabscheidung aus einem sauren KupfersulfatElektrolyten mit Polyethylenglykol und Chlorid-Zusatz bei 25 °C und natürlicher Konvektion [4]](/images/stories/Abo-2022-11/thumbnails/gt-2022-11-0044.jpg "Abb. 2 rechts: Potentiodynamische StromdichteÜberspannungskurve für die Kupferabscheidung aus einem sauren KupfersulfatElektrolyten mit Polyethylenglykol und Chlorid-Zusatz bei 25 °C und natürlicher Konvektion [4]") Fig. 2 right: Potentiodynamic current density overvoltage curve for copper deposition from an acidic copper sulphate electrolyte with polyethylene glycol and chloride addition at 25 °C and natural convection [4]
Fig. 2 right: Potentiodynamic current density overvoltage curve for copper deposition from an acidic copper sulphate electrolyte with polyethylene glycol and chloride addition at 25 °C and natural convection [4]
The steep gradients noted by Osterwald and Schulte in the potentiodynamic current density-voltage curve during copper deposition were subjected to careful analysis using the steady-state curve, overcoming difficulties of slow steady-state adjustment and distortions due to the ohmic voltage drop. Normally the steady state of the phase boundary changes continuously with the electrode voltage and accordingly the steady state current density is a continuous function of the electrode voltage. In the work in the acidic copper sulphate electrolyte, however, it was shown that the steady-state current density overvoltage curve became discontinuous and ambiguous at a characteristic overvoltage ηT and is limited at this discontinuity point by two current densities IA and IB at the same electrode potential [7] (Fig. 3). The dash-dotted extensions in the stationary curve branch a and curve branch b correspond to metastationary states. The vertical curve in the potentiodynamic characteristic, which apparently indicates that the electrode is unpolarizable, was actually due to the coexistence of two different states on the electrode surface.

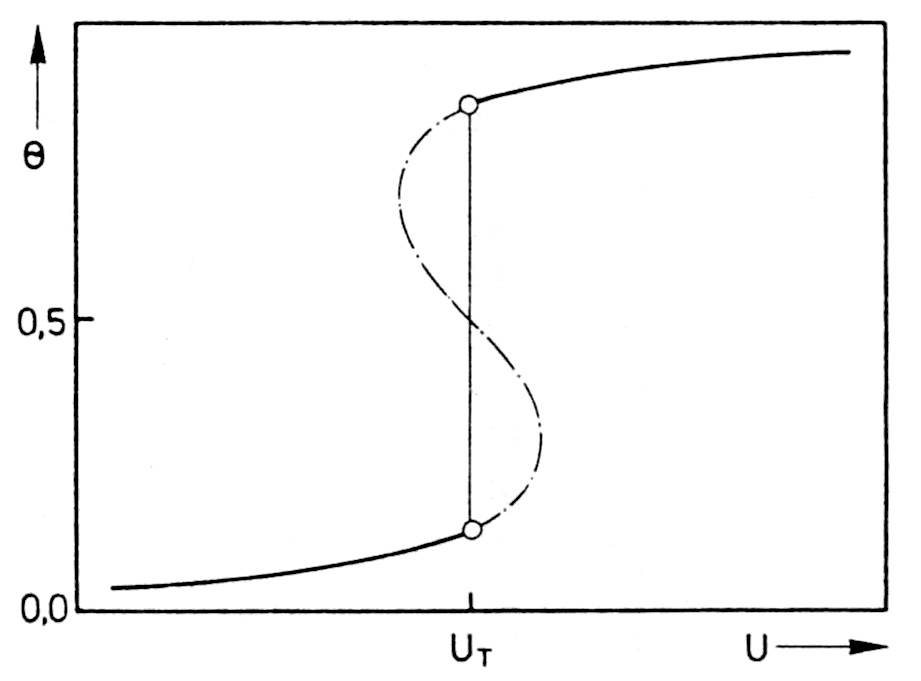
A satisfactory interpretation of this could be described in the investigated case by the adsorption behavior of the polyethylene glycol in the presence of chloride ions by Frumkin's adsorption isotherm, if sufficient attraction between the adsorbed molecules is assumed [8]. Under these circumstances, the degree of adsorption depends on the electrode voltage. The states corresponding to the S-shaped part of the U-Θ curve in Figure 4 are not stable. As a result, there is an equilibrium between two two-dimensional interfacial phases at the point UT. This means that two-dimensional condensation takes place at point UT when the electrode voltage increases and two-dimensional evaporation takes place when it decreases. Both processes lead to a sudden change in the degree of coverage.
Since the polyethylene glycol has an inhibiting effect on copper deposition in the presence of chloride, the observed discontinuity of the electrode characteristic curve is associated with the abrupt change in the degree of coverage. The initial process involves the formation of nuclei in the interfacial phase, which is comparable to the nucleation during the condensation of a gas or the evaporation of a liquid. It should be added that the coexistence of two different surface phases is possible with the same electrode voltage and the same electrolyte composition, since the current densities IA and IB (Fig. 3) are not so different that there is a drastic change in the total concentration and therefore correspond to equal conditions. Accordingly, in Fig. 3, ηT proved to be independent of the convection conditions in front of the electrode. In the case that the current density is between current densities IA and IB, which correspond to states A and B (Fig. 3), the interfacial phase cannot maintain the same state on the entire electrode surface. Instead, some areas of the surface are in state A and others in state B.
This coexistence of two different steady states of the same condition turns a flat electrode surface into an uneven surface due to the corresponding different current densities IA and IB (Fig. 5 a, b). The ratio of the different area fractions for the two surface states follows a kind of lever law, whereby their size and distribution on the electrode surface cannot be predicted, as these depend, among other variables, on the nucleation frequency according to the experimental design and the condition of the substrate. In the continuous branches a and b of the electrode characteristic curve, the metal is deposited evenly over the entire electrode, albeit with significantly different current densities.
 Zustand a überwiegt; b) Zustand b überwiegt") Fig. 5: Cathode surface with two coexisting surface states: a) State a predominates; b) State b predominates
Fig. 5: Cathode surface with two coexisting surface states: a) State a predominates; b) State b predominates
The anodic phenomenon of pitting corresponds to the formation of buds, pustules and dendrite-like structures in cathodic metal deposition as a direct result of local differences in current density. Surprisingly, this analogy was first recognized by Osterwald and his colleagues, who subsequently carried out investigations in order to gain a better understanding of pitting corrosion by elucidating the cathodic processes.
A theory of pitting corrosion has two special tasks: it must explain why areas with high and low dissolution rates can coexist on one and the same surface, and it must explain the initial process. The first studies dealing with the theory of pitting corrosion dealt with pitting on passive surfaces. It was assumed that the holes corrode in the active state and that the consequences of the high pitting current density prevent repassivation. The direct consequences include the ohmic voltage drop and the concentration shift. In fact, conductivity or mass transfer effects or both can stabilize the state of heterogeneous metal dissolution. Rickert and Holzäpfel had given convincing examples in which the inhomogeneous metal dissolution is maintained solely by diffusion processes in the Nernst layer [11]. However, following an objection by Vetter and Strehblow [13], it is difficult to understand the first phase of hole nucleation growth on the basis of conductivity and transport effects. The two authors therefore demanded that a theory of pitting corrosion must be free of such effects in its basic assumptions.
![Abb. 6 links: Schematische Darstellung einer Unstetigkeitsstelle in der anodischen Stromdichte-Spannungskennlinie [9]](/images/stories/Abo-2022-11/thumbnails/gt-2022-11-0049.jpg "Abb. 6 links: Schematische Darstellung einer Unstetigkeitsstelle in der anodischen Stromdichte-Spannungskennlinie [9]") Fig. 6 left: Schematic representation of a discontinuity in the anodic current density-voltage characteristic [9]
Fig. 6 left: Schematic representation of a discontinuity in the anodic current density-voltage characteristic [9]
![Abb. 7 rechts: Stationäre StromdichteSpannungskennlinie eines NickelEinkristalls mit [111] Orientierung in 8 N methanolischer wasserfreier Schwefelsäure bei 25 °C [9]](/images/stories/Abo-2022-11/thumbnails/gt-2022-11-0050.jpg "Abb. 7 rechts: Stationäre StromdichteSpannungskennlinie eines NickelEinkristalls mit [111] Orientierung in 8 N methanolischer wasserfreier Schwefelsäure bei 25 °C [9]") Fig. 7 right: Stationary current density-voltage characteristic of a nickel single crystal with [111] orientation in 8 N methanolic anhydrous sulphuric acid at 25 °C [9]
Fig. 7 right: Stationary current density-voltage characteristic of a nickel single crystal with [111] orientation in 8 N methanolic anhydrous sulphuric acid at 25 °C [9]
If the findings of inhomogeneous metal deposition in the cathodic part of the current density-voltage curve are transferred to the anodic metal dissolution, the phenomenon of pitting corrosion should therefore be found in the area of a discontinuity in the anodic part of the current density-voltage curve (Fig. 6). According to the theory of coexisting steady states, the initiation of pitting corrosion can fulfill all the requirements demanded by Vetter and Strehblow. The typical phenomena of pitting corrosion were to be expected when the number of sites with high dissolution current density IB is large, their area fraction is small and the differences between IA and IB are considerable (Fig. 6).
The investigation of branch b posed considerable experimental problems, which were mainly due to the high hole current density. Kaesche [12] had already pointed out this problem. To overcome this experimental hurdle, Osterwald and colleagues used a trick by deliberately amplifying the secondary effects and facilitating the formation of a salt layer (Fig. 7).
![Abb. 8: Nickel-Einkristalloberfläche nach abrupter Erhöhung der Stromdichte von der passiven Zone in den Bereich der heterogenen Metallauflösung [9]](/images/stories/Abo-2022-11/thumbnails/gt-2022-11-0051.jpg "Abb. 8: Nickel-Einkristalloberfläche nach abrupter Erhöhung der Stromdichte von der passiven Zone in den Bereich der heterogenen Metallauflösung [9]") Fig. 8: Nickel single crystal surface after abruptly increasing the current density from the passive zone to the area of heterogeneous metal dissolution [9]
Fig. 8: Nickel single crystal surface after abruptly increasing the current density from the passive zone to the area of heterogeneous metal dissolution [9]
As a result, the current densities of IB and branch b of the stationary current density-voltage characteristic could be significantly reduced. A suitable system was 8 N methanolic anhydrous sulphuric acid (21.9 Ohm cm). In this system, Osterwald and coworkers were indeed able to produce various forms of pitting corrosion on nickel single crystals [9, 16, 17]. In Figure 8, the typical pitting structure on the nickel single crystal was generated by abruptly increasing the current density from the passive zone of the current density-voltage curve into the region of heterogeneous dissolution.
Studies on zinc electrodes in the presence of interfacially active substances have shown that the formation of typical pitting phenomena according to the theory of coexisting steady states is not necessarily linked to the passive state, whereby typical pitting structures could be generated [7, 15].
Although these theories developed by Osterwald and colleagues in the 1980s had far-reaching industrial implications and still have them today, many of them have not been deeply anchored in the knowledge pool of electroplating technology, as the above-mentioned little story of non-uniform gold deposition shows, and some were even "rediscovered" many years later abroad [14]. Due to the tragic accidental death of Prof. Osterwald in March 1982, the work on inhomogeneous metal deposition and dissolution could not be continued, which, in addition to the sometimes unwelcome questioning of scientific legacies, is certainly also a reason for the repeated lack of knowledge of the electroplating knowledge already acquired.
Literature
[1] Galvanotechnik, 82, [1991] 3487-3488
[2] Fischer, H.: Electrolytic Deposition and Electrocrystallization of Metals, Springer-Verlag, Berlin, Göttingen, Heidelberg, 1954
[3] Matschke, H: Dipl. thesis, TU Berlin 1949
[4] Osterwald, J. and Schulte, H.: Galvanotechnik 67 [1976] 440-443
[5] Osterwald, J., Schulz-Harder, J.: Galvanotechnik 66 [1975] 360 - 365
[6] Landau, U., Osterwald, J.: Erzmetall 29 [1976] 103 - 109
[7] Ammon, H., Disam, J., Osterwald, J. and Schulte, H.: Werkstoffe und Korrosion 30 [1979] 690-694
[8] Damaskin, B. B., Petrii, O. A., Batrakov, V. V.: Adsorption of Organic Compounds on Electrodes, New York [1971] 85 ff
[9] Amon, H., Osterwald, J.: Proceedings of the 8 th International Congress on Metallic Corrosion, Mainz [1981]: Coexistence of Equiconditioned Steady States and Pitting Corrosion
[10] Osterwald, J.: Proc. of an International Symposium Honoring Professor H. H. Uhlig on his 75th Birthday, [1981], Denver, Colorado 112-119
[11] Rickert, H. and Holzapfel, G.: Werkstoffe und Korrosion, 17, Heft 5, [1966] 376-380
[12] Kaesche, H.: Corrosion of metals, Berlin [1966] p. 276
[13] Vetter, K. J., Strehblow, H. H.: Ber. Bunsenges. 74 [1970] 449-455
[14] Landau, U.: Galvanotechnik 106 [2015] 1176 - 1179
[15] Disam, J.: Diss. TU Berlin [1979]
[16] Steppke, D.: Diss. TU Berlin [1978]
[17] Amon, H.: Diss. TU Berlin [1982]
[18] Schulte, H.: Diss. TU Berlin [1976]
[19] Landau, U.: Diss. TU Berlin [1976]
[20] Schulz-Harder, J.: Diss. TU Berlin [1971]
